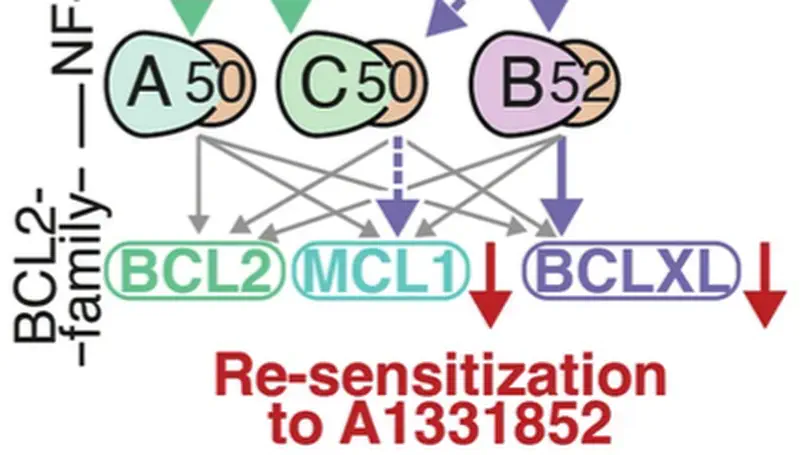
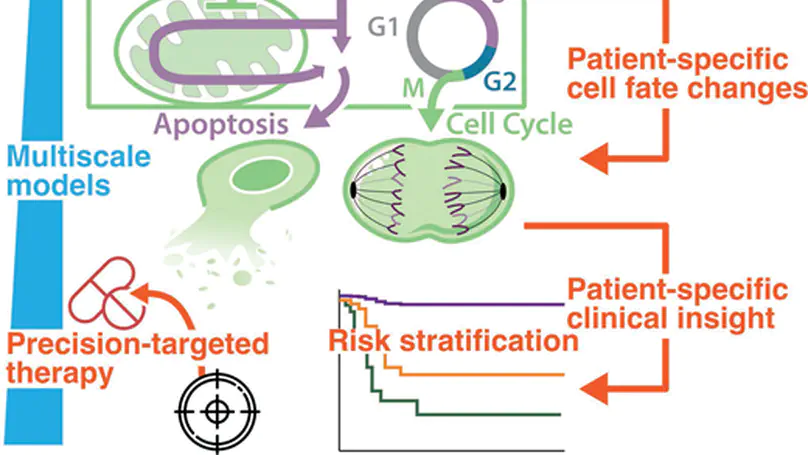
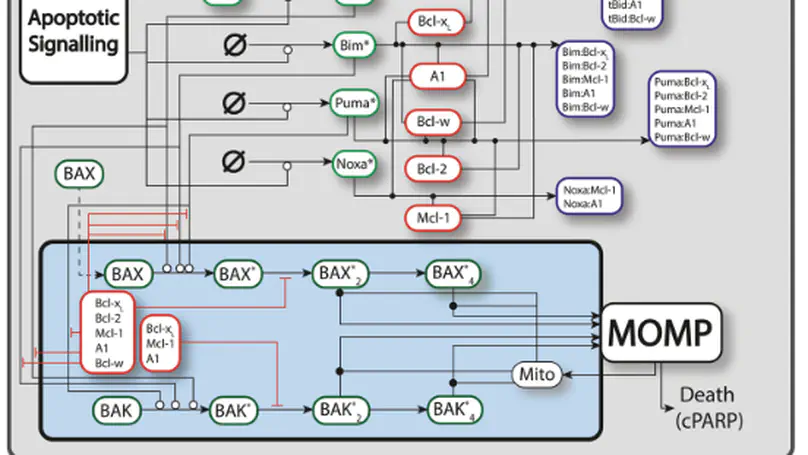
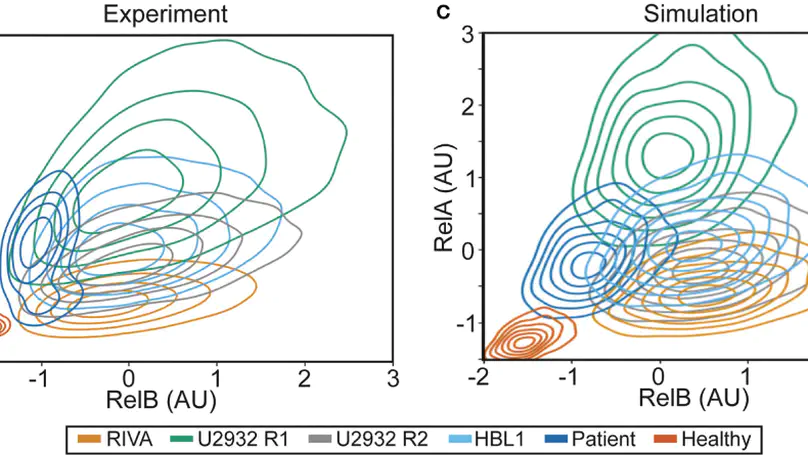
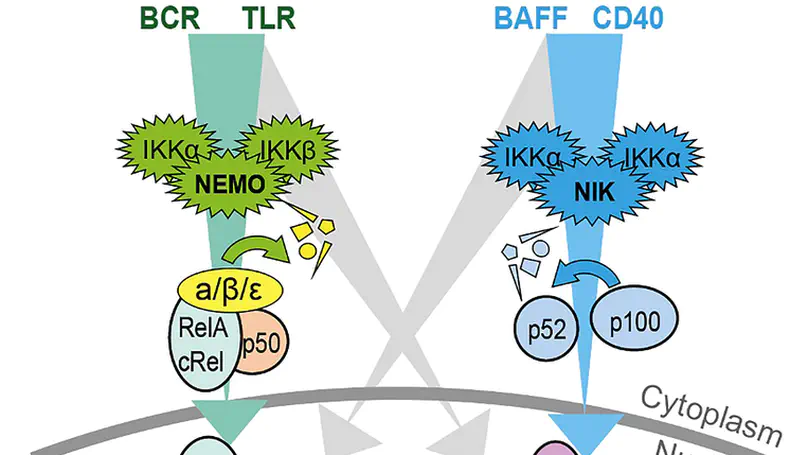
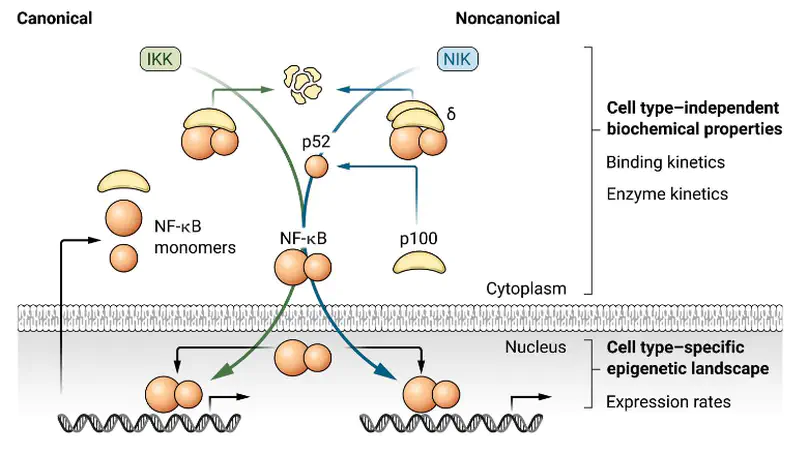
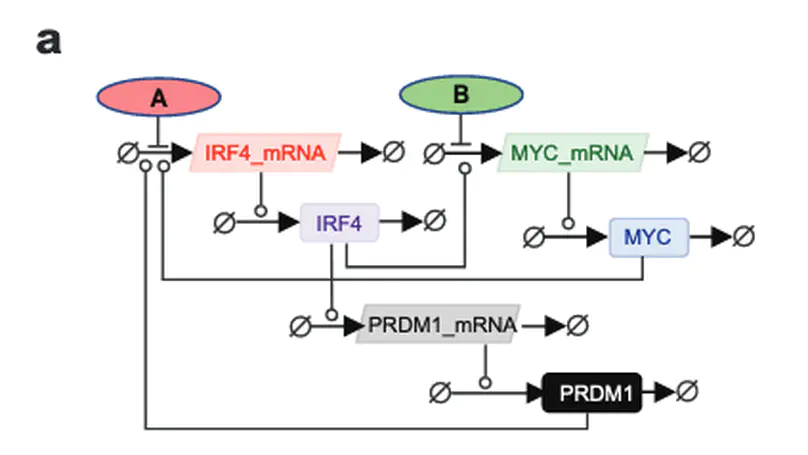
In Diffuse Large B-cell Lymphoma (DLBCL), elevated anti-apoptotic BCL2-family proteins (e.g., MCL1, BCL2, BCLXL) and NF-κB subunits (RelA, RelB, cRel) confer poor prognosis. Heterogeneous expression, regulatory complexity, and redundancy offsetting the inhibition of individual proteins, complicate the assignment of targeted therapy. We combined flow cytometry ‘fingerprinting’, immunofluorescence imaging, and computational modeling to identify therapeutic vulnerabilities in DLBCL. The combined workflow predicted selective responses to BCL2 inhibition (venetoclax) and non-canonical NF-κB inhibition (Amgen16). Within the U2932 cell line we identified distinct resistance mechanisms to BCL2 inhibition in cellular sub-populations recapitulating intratumoral heterogeneity. Co-cultures with CD40L-expressing stromal cells, mimicking the tumor microenvironment (TME), induced resistance to BCL2 and BCLXL targeting BH3-mimetics via cell-type specific upregulation of BCLXL or MCL1. Computational models, validated experimentally, showed that basal NF-κB activation determined whether CD40 activation drove BH3-mimetic resistance through upregulation of RelB and BCLXL, or cRel and MCL1. High basal NF-κB activity could be overcome by inhibiting BTK to resensitize cells to BH3-mimetics in CD40L co-culture. Importantly, non-canonical NF-κB inhibition overcame heterogeneous compensatory BCL2 upregulation, restoring sensitivity to both BCL2- and BCLXL-targeting BH3-mimetics. Combined molecular fingerprinting and computational modelling provides a strategy for the precision use of BH3-mimetics and NF-κB inhibitors in DLBCL.