Welcome to the Mitchell Lab
At Brighton and Sussex Medical School, The University of Sussex
We’re tackling cancer with systems biology, in the beautiful city of Brighton, UK.
What we do
Our interdisciplinary approach aims to bring systems biology approaches to the clinic.
Mathematics
to represent cell signaling
Computing
to simulate disease
Experiments
to test predictions
Medicine
to improve treatments
Latest News




Projects
*
Meet the Team
Principal Investigator
Simon Mitchell
Professor in Cancer Research
Postdoctoral Researchers
Aimilia Vareli
Postdoctoral Researcher
Tabea Fullstone
Postdoctoral Researcher
PhD Students
Abigail Edwards
PhD Student
Isabelle Jupp
PhD Student
Administration
Gemma Hamilton
Administration
Lab Alumni
Featured Publications
Recent Publications
(2025).
Development of a translational strategy for using TIMP-3 to inhibit aggrecanase activity in osteoarthritis.
Osteoarthr. Cartil..
(2025).
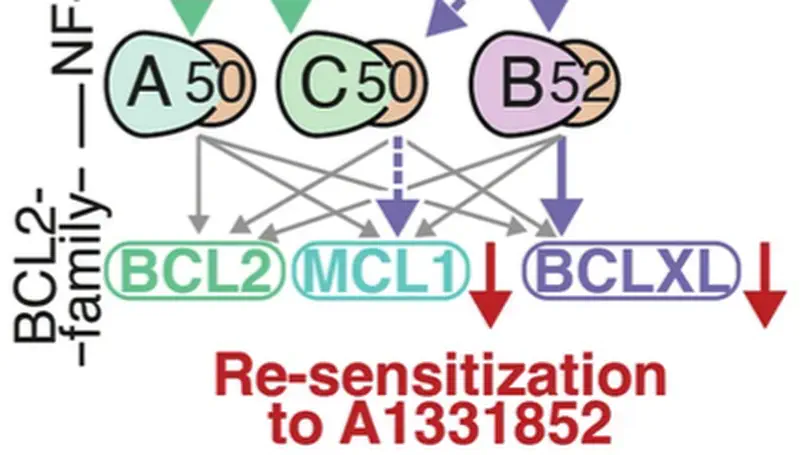
Systems biology-enabled targeting of NF-κΒ and BCL2 overcomes microenvironment-mediated BH3-mimetic resistance in DLBCL.
Cell Death Dis.
(2025).
Computational modelling of aggressive B-cell lymphoma.
Biochem Soc Trans.
(2025).
Design, synthesis and evaluation of Pyrrolobenzodiazepine (PBD)-based PROTAC conjugates for the selective degradation of the NF-κB RelA/p65 subunit.
RSC Medicinal Chemistry.
(2025).
A Systems Biology Approach of Quantifying Signal Transduction to B-Cell Proliferation and Differentiation.
B-Cell Receptor Signaling.
Contact
- S.A.Mitchell@bsms.ac.uk
- Medical Research Building, University of Sussex, Falmer, East Sussex, BN1 9PX
- Enter the Medical Research Building (note this is behind the Medical Teaching Building) and call reception. We are on the second floor, up the stairs through the security door.
- Institution Website
- +44 (0)1273 678584
- ORCID iD
- Book a Meeting with Simon
- Google Scholar