Abstract
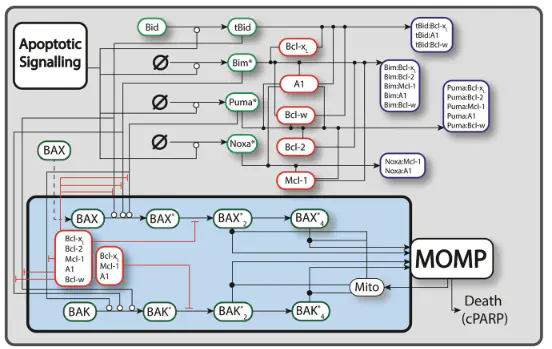
In healthy cells, pro- and anti-apoptotic BCL2 family and BH3-only proteins are expressed in a delicate equilibrium. In contrast, this homeostasis is frequently perturbed in cancer cells due to the overexpression of anti-apoptotic BCL2 family proteins. Variability in the expression and sequestration of these proteins in Diffuse Large B cell Lymphoma (DLBCL) likely contributes to variability in response to BH3-mimetics. Successful deployment of BH3-mimetics in DLBCL requires reliable predictions of which lymphoma cells will respond. Here we show that a computational systems biology approach enables accurate prediction of the sensitivity of DLBCL cells to BH3-mimetics. We found that fractional killing of DLBCL, can be explained by cell-to-cell variability in the molecular abundances of signaling proteins. Importantly, by combining protein interaction data with a knowledge of genetic lesions in DLBCL cells, our in silico models accurately predict in vitro response to BH3-mimetics. Furthermore, through virtual DLBCL cells we predict synergistic combinations of BH3-mimetics, which we then experimentally validated. These results show that computational systems biology models of apoptotic signaling, when constrained by experimental data, can facilitate the rational assignment of efficacious targeted inhibitors in B cell malignancies, paving the way for development of more personalized approaches to treatment.